







ARTICLES
专业文章
ICH视角下2020版中国GCP简评及申办者合规管理
ICH视角下2020版中国GCP简评及申办者合规管理
2020年4月26日,国家药品监督管理局与国家卫健委联合发布新版《药物临床试验质量管理规范》("《规范》"),并明确于2020年7月1日施行。《规范》分为"术语及其定义"、"伦理委员会"、"研究者"、"申办者"、"试验方案"、"研究者手册"等章节,分别从伦理委员会、研究者、申办者等维度体现其各自在药物临床试验中的职责。
自2017年6月1日起,中国正式成为ICH的成员国。为适应ICH,国家市场监督管理总局于2018年公布了《药物临床试验质量管理规范(修订草案征求意见稿)》,吸收了ICH E6 R2的大部分内容。在2003版《药物临床试验质量管理规范》("旧版《规范》")的基础上,最新版《规范》结合中国临床的实际情况作出了进一步的调整和优化,其整体框架与ICH-GCP一致,但在部分细节上稍有差异。
本文将比对ICH-GCP和《规范》的主要条款,并从《规范》对申办者的要求角度入手,结合近期新出台的其他重要法律法规,为申办者快速总结其在药物临床试验过程中的主要合规管理要点和职责。
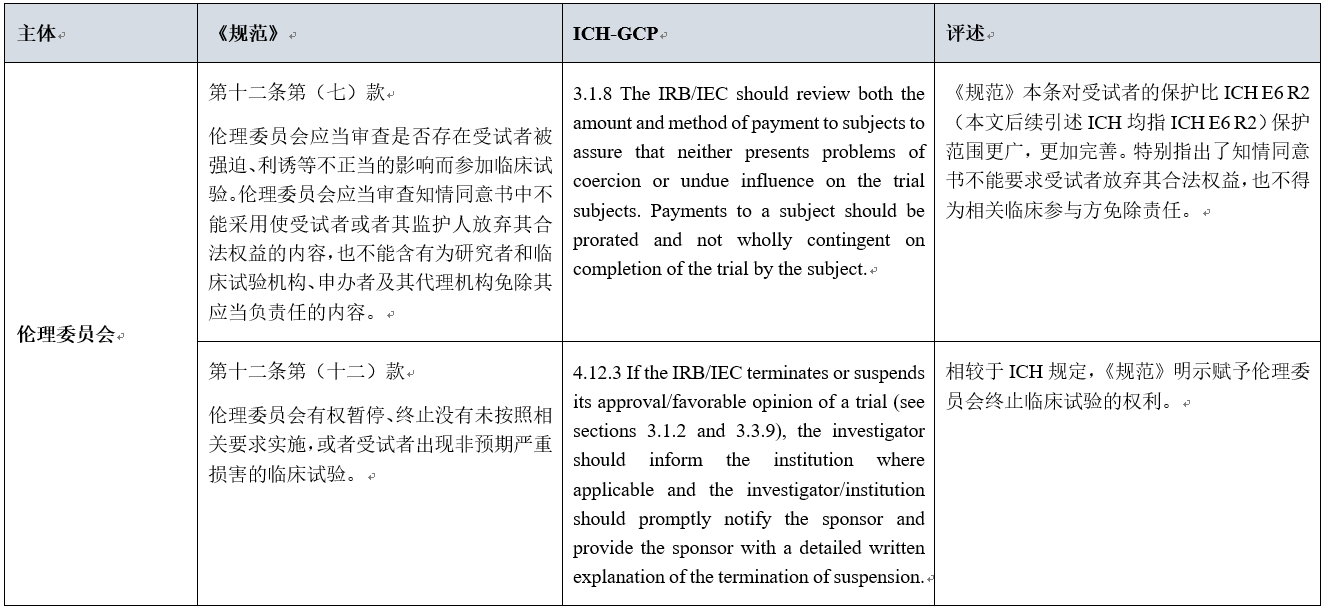
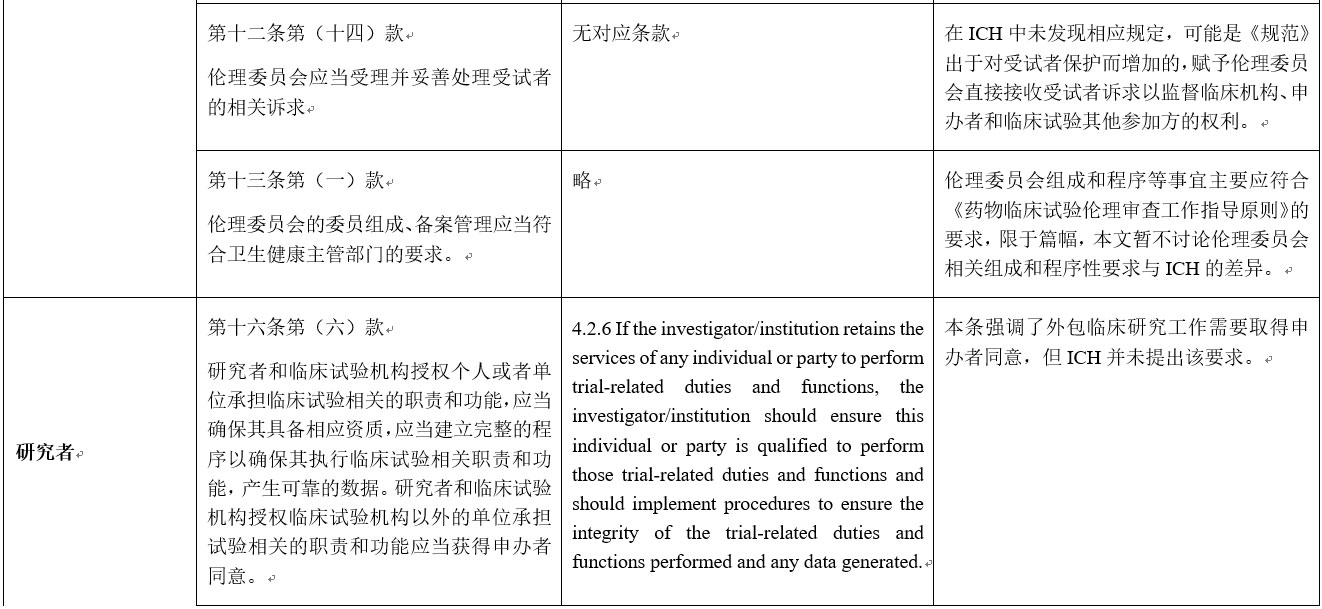


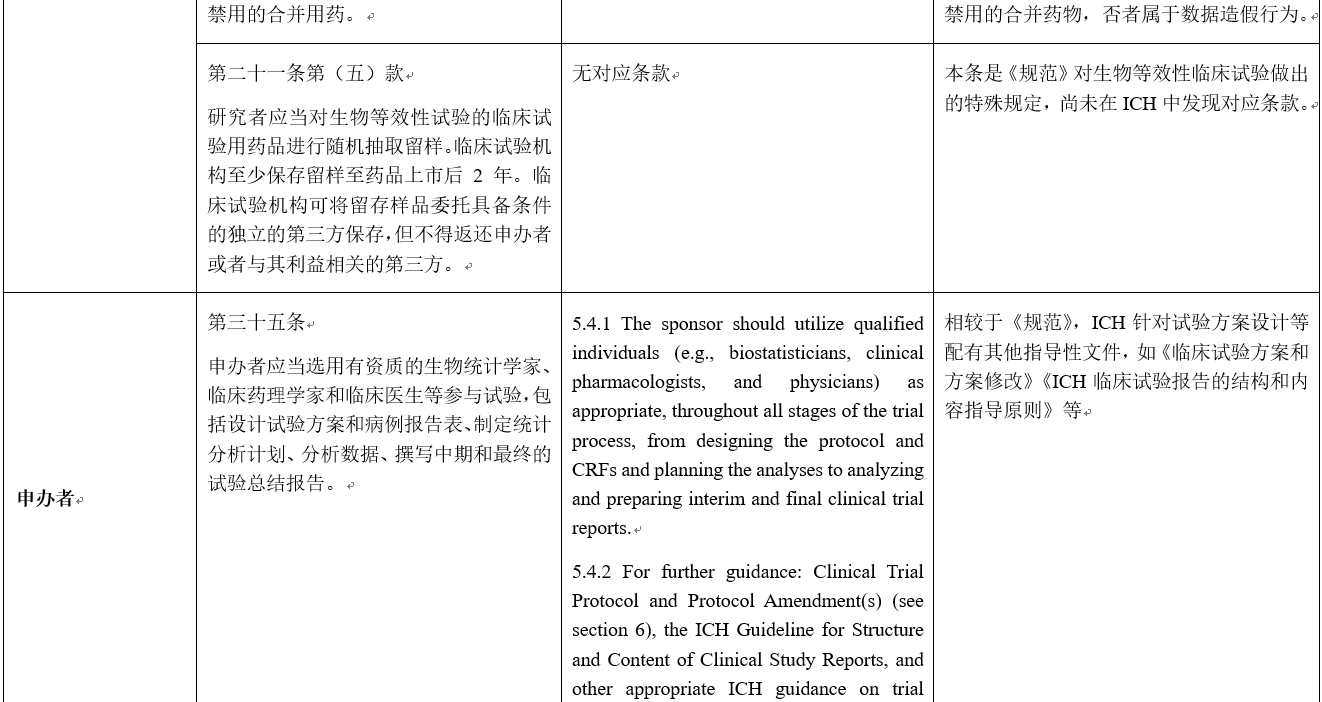


点击图片可查看大图
申办者是临床合规的主要责任方
在药物临床试验法律关系中,受试者同时与申办者、研究机构建立药物临床试验合同关系。申办者在临床试验各方参与临床试验前与各方签订合同,明确其职责,是《规范》的强制性规定。申办者委托研究者或合同研究组织(CRO)开展药物临床试验是委托与被委托的法律关系,双方签订的合同属于委托合同。虽然,《国家食品药品监督管理总局关于进一步做好药物临床试验数据自查核查工作有关事宜的公告》(2015年第166号)已明确药物临床试验申请人和药品注册申请人对临床试验数据真实性承担全部法律责任,但此仅为对外的责任承担形式,因研究者或CRO的过失给申办者造成损害的,申办者仍然可以依据合同要求其承担赔偿责任。
根据《规范》第四十条的规定,申办者与研究者和CRO签订的合同,应当明确试验各方的责任、权利和利益,以及各方应当避免的、可能的利益冲突。合同内容中应当包括:临床试验的实施过程中遵守相关的临床试验的法律法规;执行经过申办者和研究者协商确定的、伦理委员会同意的试验方案;遵守数据记录和报告程序;同意监查、稽查和检查;临床试验相关必备文件的保存及其期限;发表文章、知识产权等的约定。此外,针对研究者和CRO,我们建议分别做以下针对性的约定:
(1)申办者与研究者
由于《规范》要求申办者在选择研究者时要符合一定的要求,申办者除了在甄别研究者时应当慎重之外,还可以要求研究者在签署合同时对自身的资质做一定的保证,例如:
研究者应当经过临床试验的培训、有临床试验的经验,有足够的医疗资源完成临床试验;
涉及医学判断的样本检测实验室,应当符合相关规定并具备相应资质。临床试验中采集标本的管理、检测、运输和储存应当保证质量。禁止实施与伦理委员会同意的试验方案无关的生物样本检测(如基因等)。临床试验结束后,剩余标本的继续保存或者将来可能被使用等情况,应当由受试者签署知情同意书,并说明保存的时间和数据的保密性问题,以及在何种情况下数据和样本可以和其他研究者共享等。
(2)申办者与CRO
《规范》申办者委托给CRO的工作应当签订合同。合同中应当明确以下内容:委托的具体工作以及相应的标准操作规程;申办者有权确认被委托工作执行标准操作规程的情况;对被委托方的书面要求;被委托方需要提交给申办者的报告要求;与受试者的损害赔偿措施相关的事项;其他与委托工作有关的事项。CRO如存在任务转包,应当获得申办者的书面批准。
此外,申办者可以要求CRO在签署合同时对自身的资质及行为做一定的保证,例如:CRO应当实施质量保证和质量控制。
《规范》对申办者的要求也同样应当适用于CRO,因此,申办者与CRO签订合同时,需要考虑与CRO之间的责权利平衡。
受试者、研究者的补偿及赔偿
《规范》指出,申办者应当采取适当方式保证可以给予受试者和研究者补偿或者赔偿;申办者应当向研究者和临床试验机构提供与临床试验相关的法律上、经济上的保险或者保证。
根据《中华人民共和国侵权责任法》第五十四条,患者在诊疗活动中受到损害,医疗机构及其医务人员有过错的,由医疗机构承担赔偿责任。一般而言,对于发生与试验相关的损害或者死亡的受试者承担治疗费用及法律规定的经济补偿,在临床试验机构及研究者不存在过错的情况下,均由申办者承担。因此,申办者在与研究者签署的临床试验协议中,明确试验中的职责分工以及受试者不良反应的处理原则等就显得尤为重要。
附录:《规范》及ICH-GCP对应条款清单
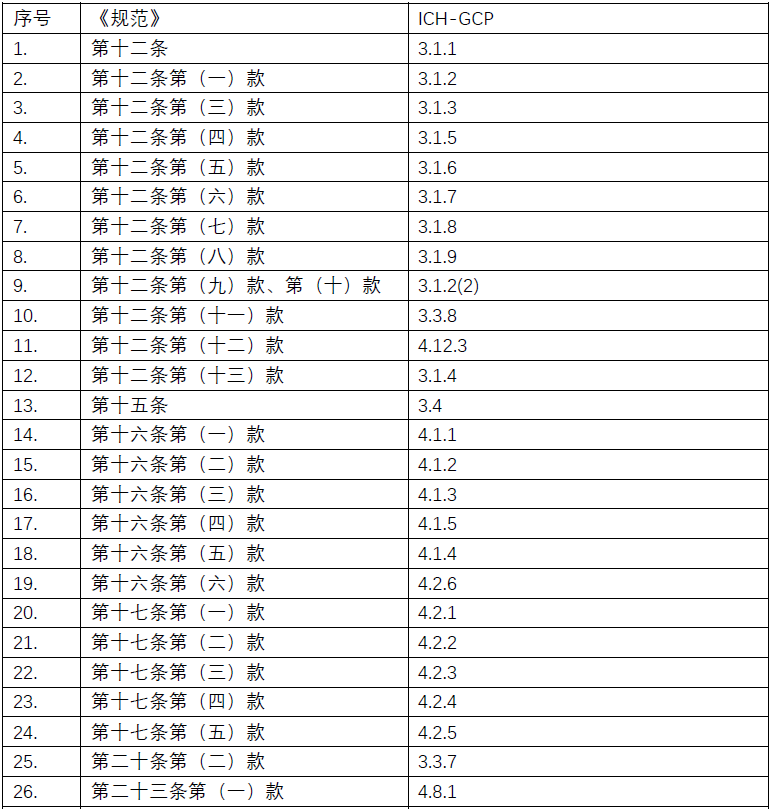
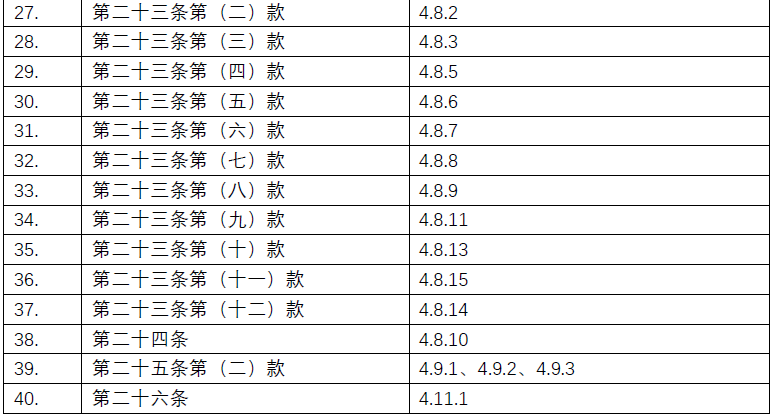
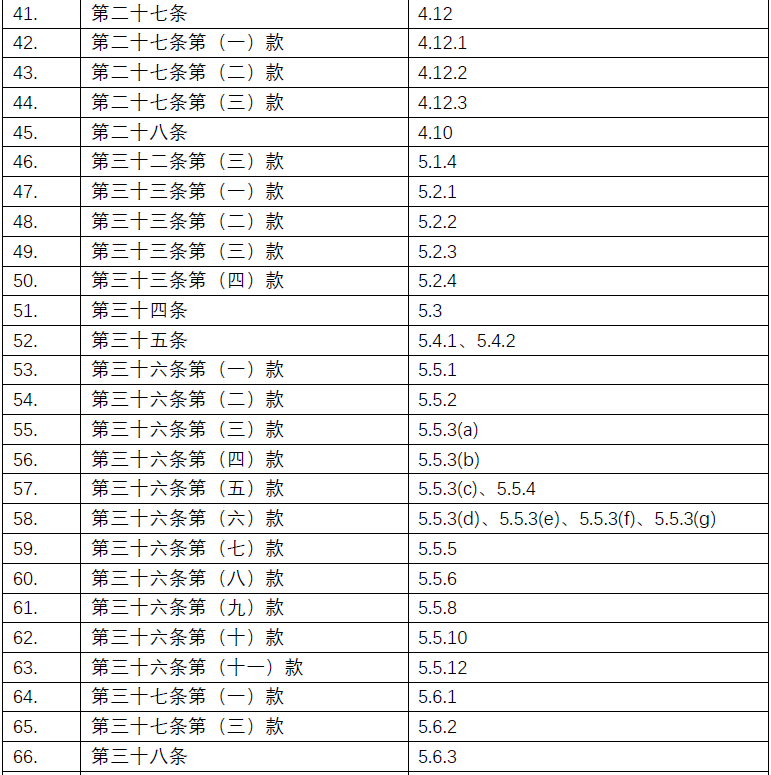
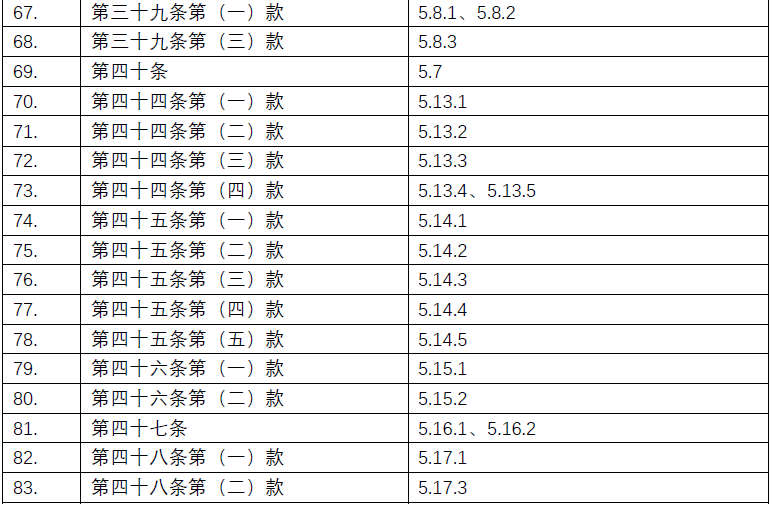
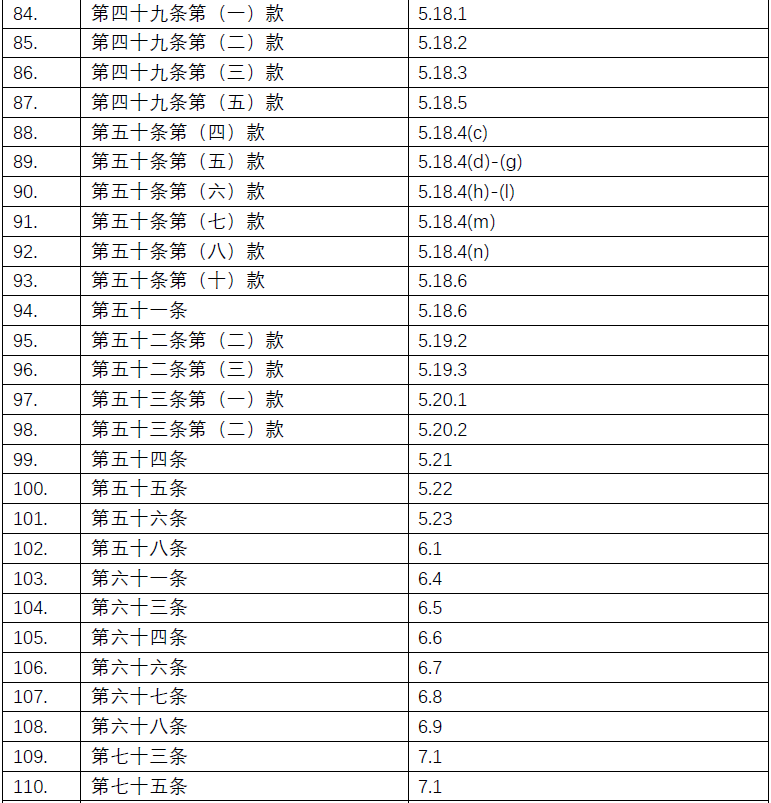

点击图片可查看大图

